Skip to content
#ReadPaper #DeepLearning #Bioinfo #ProteinStructurePrediction #AlphaFold2 #Transformer
AlphaFold2
+ [Highly accurate protein structure prediction with AlphaFold](https://www.nature.com/articles/s41586-021-03819-2)
- [[AlphaFold2]] 被 《Science》 评选为2021年AI在科学界最大的突破
- [[AlphaFold2]] 把通过氨基酸序列对蛋白质三维结构预测的误差,降低到原子级别
Model Architecture
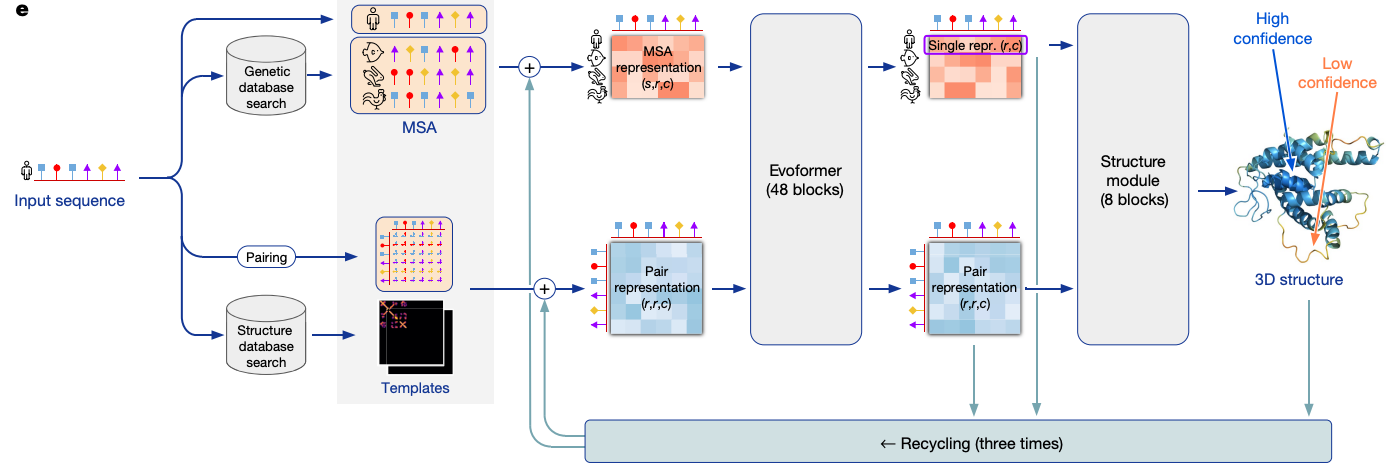
模型大概可以分成四个部分
- Feature Extraction
- Encoder
- Decoder
- Recycling
Feature Extraction
特征提取两部分信息
- MSA
- Pairing & Templates
MSA (Multi-Sequence-Alignment)
MSA提取序列在多物种中的共进化信息
- 在基因数据库中搜索相似的氨基酸序列
- 多序列比对
- 得到不同氨基酸序列的特征
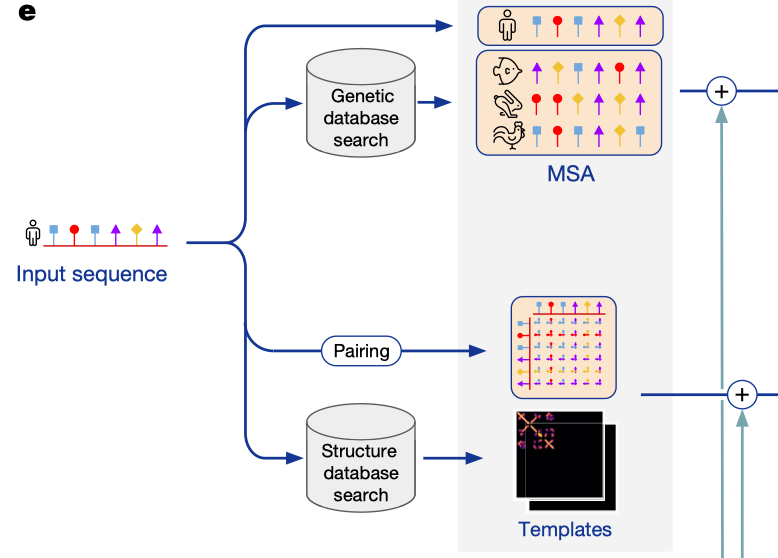
Pairing & Templates
提取氨基酸之间的特征
- 氨基酸之间的关系
- 在结构数据库中搜索,从已知的氨基酸序列的结构中搜索得到空间信息
Encoder
Encoder Input
MSA Representation
- s: s 行氨基酸序列
- 第一行是人的氨基酸序列
- 后面s-1 是从基因数据库中匹配到的不同物种的氨基酸序列
- r: r个氨基酸
- c: 每个氨基酸表示为长为c的向量
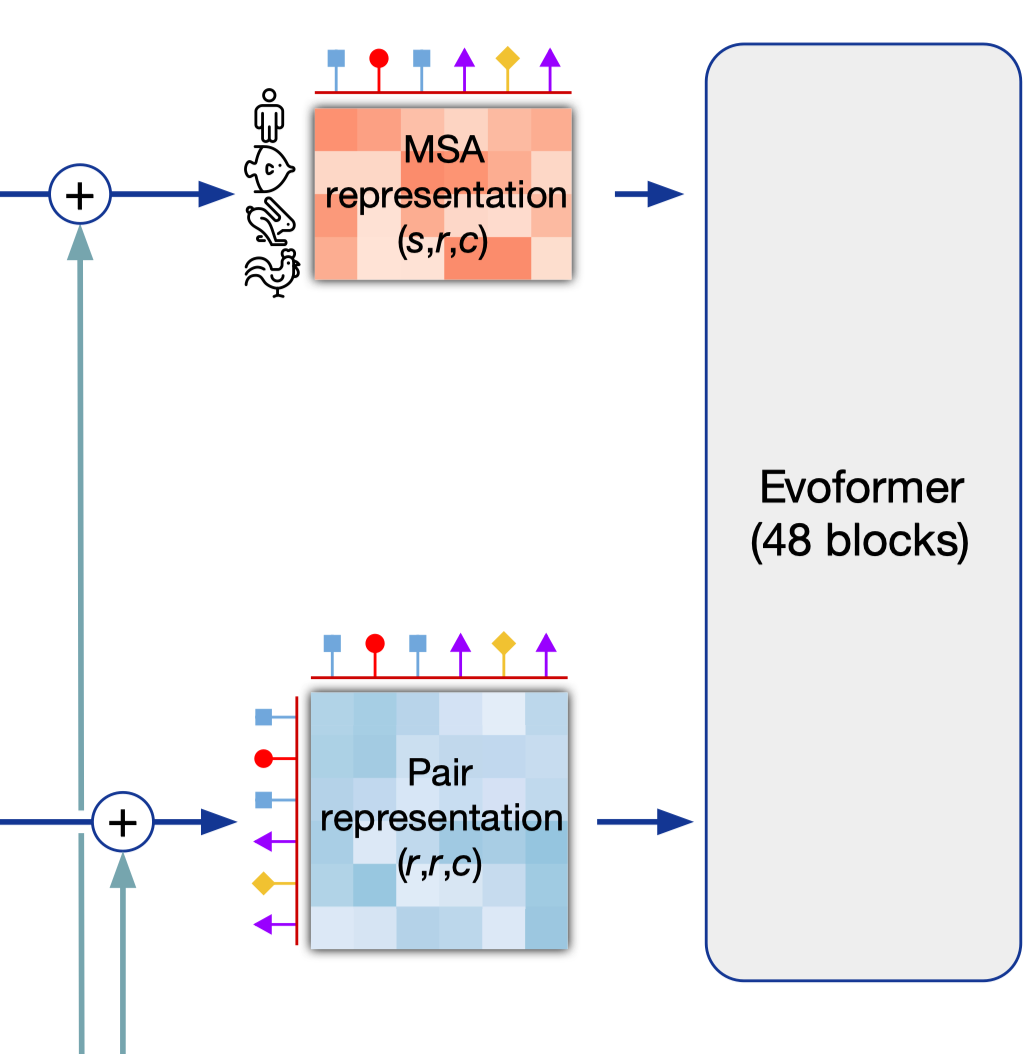
Pair Representation
- r: r个氨基酸
- c: 每个氨基酸表示为长为c的向量
- 主要用于表示氨基酸两两之间的空间信息
Evoformer Block
- 与Transformer 不同
- Transformer 输入的是一段序列
- Evoformer 在这里输入的是一个二维的矩阵
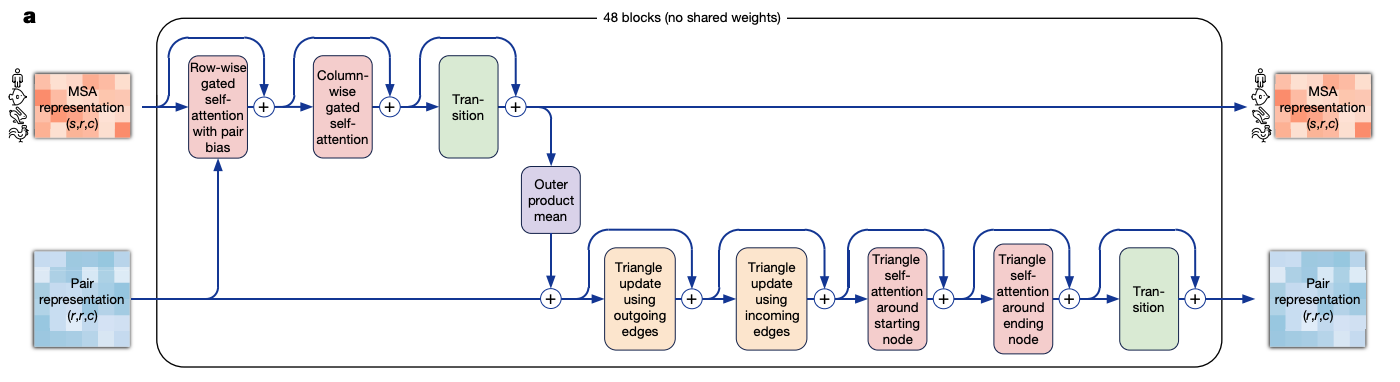
MSA和Pair都会进入多头自注意力模块
- 具有残差连接和在元素作用上的MLP
Pair的部分还需要加入物理上三角不等式的处理
与Transformer不同
- 氨基酸对 (Pair Representation) 之间的信息会加入对序列的建模
- 序列建模之后也会加入对氨基酸对的建模
- 自注意力机制做两次
- 因为输入是一个二维的矩阵
- 因为输入是一个二维的矩阵
Encoder Output
- 输出的矩阵维度不发生变化, c发生变化
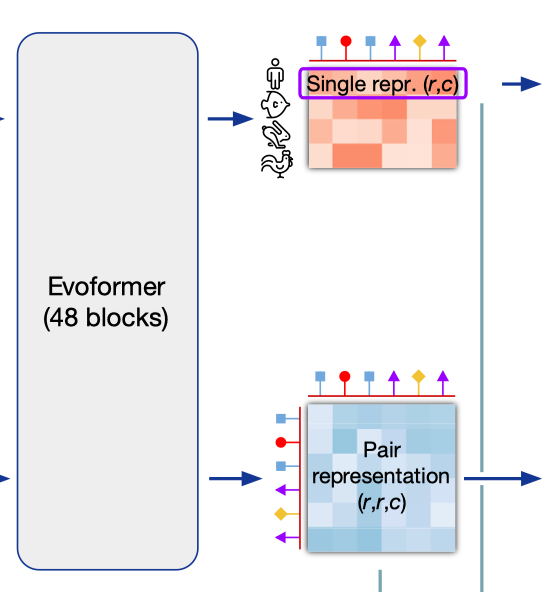
Decoder
根据Encoder的输出拿到
- Single Representation (r, c) 人氨基酸序列
- 人类氨基酸所有的特征的表示
- Pair Representation (r, r, c) 氨基酸对
- 氨基酸之间的相关信息
- Backbone frames
- 氨基酸序列主干的空间结构
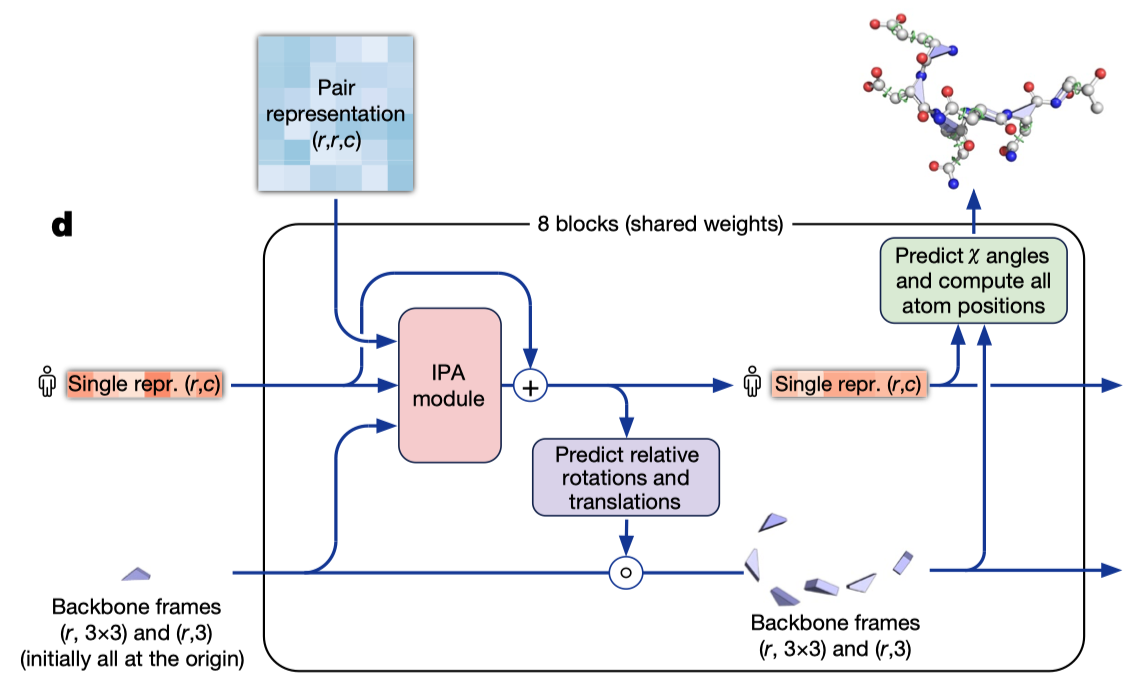
Protein 3D Structure Prediction
记录下一个氨基酸与上一个氨基酸的相对位置
- R基部分原子的旋转的角度
- 欧几里得变换
- 极性,电荷可以限制 A, B 值
- 先预测backbone上氨基酸的相对位置
- 再预测每个氨基酸R基上的原子的相对位置
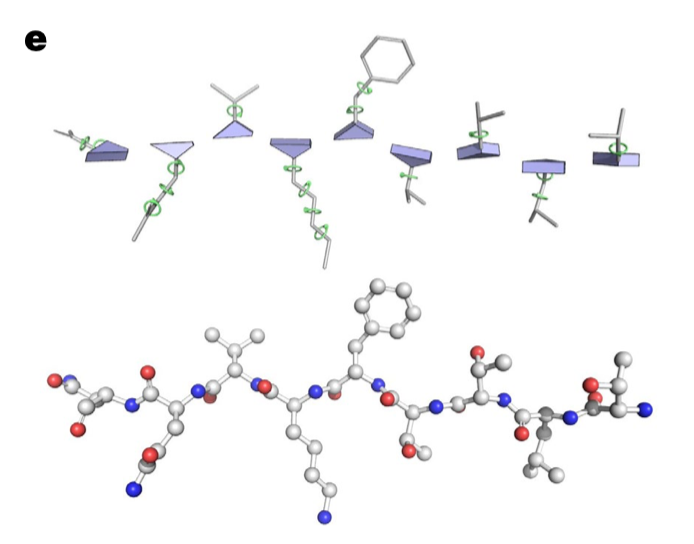
Recycling
把编码器的输出和解码器的输出通过回收机制传回编码器的输入
- 类似RNN,但是这里的梯度不反传, 只经过56层就可以计算梯度
Loss Function
Training
- 特征对齐损失,用于量化模型预测的原子位置与实际原子位置之间的差异。
- 辅助损失,它可能与多个预测任务相关,例如二级结构预测、溶剂可及性预测等。
- 距离损失,它衡量模型预测的残基间距离与真实结构之间的误差。
Training
- 多序列比对损失,用于确保模型预测与进化信息一致。
- 构象损失,用于确保整体蛋白质构象的准确性。
Fine-tuning
- 用于处理实验解析度的损失,用于在蛋白质结构中考虑到实验数据的不确定性。
- 违反物理或生物化学约束的损失。
Training with labelled and unlabelled data
Noisy student self-distillation
先在有标号的数据集PDB里训练一个模型, 通过这个模型预测没有标号的数据形成一个大一些的数据集,然后选择置信度较高的数据,加上原来有标号的数据,训练新的模型
- Using an approach similar to noisy student self-distillation.
- In this procedure, we use a trained network to predict the structure of around 350,000 diverse sequences from
Uniclust3036and make a new dataset of predicted structures filtered to a high-confidence subset.