Bioinfo Matrix Operations
Matrix Factorization
Non-negative dataset
- Counters: 计数
- Quantities: 数量数据
- Intensities: 强度数据
NMF
NMF is used in a compositional model. Data are assumed to be non-negative
Every data vector is explained as a purely constructive linear composition of a set of bases
- The bases
are in the same domain as the data
Constructive composition: no subtraction allowed
- Weights
must be non-negative - All components of bases
must be non-negative
Data Does Not Need to Be Non-Negative:
- Unlike traditional NMF where data is assumed to be non-negative, this generalization allows data to exist in any quadrant, including negative regions.
Non-Negativity Constraint:
Only the weights must remain non-negative, regardless of the data’s distribution.
The model preserves the fundamental principle of constructive addition without subtracting components.
Minimizing Divergence
Divergence: Divergence:
Iterative solution:
- Subscript
indicates thresholding -values to 0.表示对负值进行阈值处理,将所有负值设置为 0,确保非负性。
NMF is not unique
NMF is not hierarchical (非负矩阵分解并非分层结构)
- Rank-1 NMF
Rank-1 分解:将数据矩阵分解为单一基向量和权重向量的乘积。
左侧的原始矩阵被近似分解为右侧的基矩阵和权重矩阵。
这种低秩分解可以很好地捕捉数据的主成分,但可能忽略细节。
- Rank-2 NMF
Rank-2 分解:将数据矩阵分解为两个基向量和对应权重的组合。
分解为两个矩阵之和:
数据矩阵通过两个基成分加权求和表示。
Rank-2 分解能够捕捉更多的结构信息,但分解结果与 Rank-1 不具有分层关系。
非分层结构:
- 增加秩 并不会简单地在前一级结果的基础上扩展,而是需要重新计算分解。
秩的选择影响分解的质量、稀疏性和可解释性。
- 稀疏性(Sparsity):较低秩分解更稀疏,较高秩分解捕捉更多细节。
- 可解释性(Interpretability):秩的选择会影响基向量的可解释性。
- 统计保真度(Statistical Fidelity):较高秩通常能够更好地重构数据,但可能过拟合。
- 最佳秩 k 的选择: 并无统一的理论方法,需要通过实验来确定最优秩。
NMF General framework
Gradient descent generally slow
Stochastic gradient descent inappropriate
Key Approach: Alternating Minimization
- Pick starting point L₀ and R₀
- while not converged do
3. Keep R fixed, optimize L
4. Keep L fixed, optimize R
5. end while
Update steps 3 and 4 are easier than solving the full problem.
This approach is also called alternating projections or (block) coordinate descent.
Starting Point Options
- Random
- Multi-start initialization: Try multiple random starting points, run a few epochs, continue with the best.
- Based on SVD
- ...
NMF in Bioinformatics
Example - 1
非负矩阵分解(NMF) 来识别癌症基因组数据中的多维模块,通过综合分析多平台基因组数据实现: link
- DNA 甲基化(DM)
- miRNA 表达(ME)
- 基因表达(GE)
The Joint NMF Framework for Integrative Analysis
Example - 2
本示例展示了如何使用 种子非负矩阵分解(Seeded NMF) 来解析空间转录组学(Spatial Transcriptomics, ST)数据。
- 空间转录组学(ST)技术:
- 生成基因表达图谱,同时保留组织的空间上下文。
- 每个空间点(spots)通常捕获多个细胞,缺乏单细胞分辨率。
- 目标:
- 通过集成单细胞 RNA 测序(scRNA-seq)数据和 ST 数据,使用 NMF 进行去卷积(Deconvolution)。
- 解释每个空间点的细胞组成。
SPOTlight
SPOTlight 使用种子 NMF 回归(Seeded NMF Regression) 来实现目标:
学习单细胞数据中的主题特征(signatures):
- 使用单细胞 RNA 测序数据,识别不同细胞类型的基因表达特征。
解析空间数据:
- 将这些特征应用于 ST 数据,找到每个空间点的最佳加权细胞类型组合。
NMF 分解过程
初始化 (NMF Initialization)
数据分解为:
- W(基矩阵):基因 × 主题(gene × topic)。
- H(系数矩阵):主题 × 细胞(topic × cell)。
- V:基因 × 细胞的原始表达数据矩阵。
分解步骤 (Factorization)
将 ST 数据 V′V' 分解为基矩阵 WW 和系数矩阵 HH:
- WW:基因 × 主题矩阵(与单细胞特征关联)。
- HH:主题 × 空间点的权重矩阵。
解析结果:Spot 去卷积
- NNLS(非负最小二乘法):
- 用于优化权重矩阵 HH,以解释每个空间点的细胞组成。
- 结果展示:
- 每个空间点(Spot 1、Spot 2 等)分解为多个细胞类型的加权组合,显示在饼图中。
- 例如:
- Spot 1: 17% 细胞类型 A,17% 细胞类型 B,33% 细胞类型 C 等。
- 种子 NMF 的优势:
- 使用单细胞数据作为“种子”初始化 WW,提高结果的生物学可解释性。
- 空间数据去卷积:
- SPOTlight 将单细胞数据与空间数据结合,揭示每个空间点的细胞组成。
- 应用场景:
- 理解组织中的空间细胞异质性,特别在癌症等研究中具有重要意义。
总结:通过种子 NMF 回归方法,SPOTlight 有效解析空间转录组学数据,结合单细胞数据揭示组织的细胞组成。
Example - 3
背景:人类癌症中的突变特征
目标:通过非负矩阵分解(NMF)方法,揭示不同癌症样本中的突变特征(mutational signatures)。 数据:
- 横轴:癌症样本(不同的癌症类型)。
- 纵轴:突变特征(SBS1、SBS2、SBS3 等)。
- 点的大小与颜色:代表不同特征在特定癌症样本中的突变贡献程度。
主要内容解析
- 横轴(Cancer Samples):
- 不同的癌症类型(如乳腺癌、肺癌等)。
- 每一列表示一个癌症样本集合。
- 纵轴(Mutation Signatures):
- SBS(Single Base Substitution)特征:代表单碱基替换的不同突变模式(SBS1、SBS2 等)。
- 每一行代表一种特定的突变特征。
- 点的表示:
- 颜色:不同颜色表示特征的突变机制,例如:
- 橙色:APOBEC 活性
- 紫色:DNA 修复缺陷
- 蓝色:化学暴露或辐射
- 点的大小:表示特定突变特征在癌症样本中的贡献比例。
- 颜色深浅:表明突变数量的多少,越深表示突变数量越高。
- 颜色:不同颜色表示特征的突变机制,例如:
- 右侧图例(Proposed Aetiology):
- 列出了每个突变特征背后的潜在机制,例如:
- SBS1:5-甲基胞嘧啶脱氨
- SBS3:BRCA1/2 突变导致的 DNA 修复缺陷
- SBS4:与烟草吸烟相关
- 列出了每个突变特征背后的潜在机制,例如:
NMF 的应用
- 非负矩阵分解(NMF):
- 将突变数据矩阵分解为:
- X:突变数据矩阵(癌症样本 × 突变特征)。
- W:基因组突变的特征矩阵(表示不同特征的突变机制)。
- H:样本中的权重矩阵(表示各特征在不同样本中的贡献)。
- NMF 的作用:
- 提取癌症样本中的突变特征。
- 识别不同癌症类型中的主要突变机制。
- 将突变数据矩阵分解为:
关键结果
突变特征(SBS)与癌症类型的关联:
- 某些突变特征(如 SBS3)与DNA 修复缺陷相关(例如 BRCA1/2 突变)。
- SBS4 明显与烟草吸烟相关。
- 特定癌症类型的突变模式与特定环境因素或基因缺陷有关。
数据可视化:
- 点图展示了不同癌症样本中各突变特征的贡献大小。
- 可直观地识别出哪些突变机制在某些癌症类型中占主导地位。
突变机制:
- 提供了癌症突变的潜在生物学机制(如化学暴露、DNA 修复缺陷等),对癌症的病因研究具有重要意义。
总结
通过 NMF 分解:
- 揭示了人类癌症中的突变特征及其贡献程度。
- 帮助识别不同癌症类型中的主要突变机制(例如化学暴露、DNA 修复缺陷等)。
- 为癌症研究提供了一个整合、系统化的框架,揭示突变数据中的潜在模式和生物学机制。
Matrix Imputation
- Proteomics
- Single cell RNA-seq
- Spatial Transcriptomics
Proteomics
在蛋白质组学(Proteomics)数据分析中,缺失值是常见问题,解决缺失值的主要方法可以分为以下几类:
Single Value Replacement
单值替换
- 半最小值法(Half Minimum)
- 将缺失值替换为所有观测数据中最小值的一半。
- 适用场景:MNAR(Missing Not At Random,非随机缺失)。
- 均值替换(Mean)
- 将缺失值替换为所有观测值的平均值。
- 适用场景:MCAR/MAR(随机缺失/完全随机缺失)。
Global Structure Low-Rank Matrix Factorization
全局结构低秩矩阵分解
- PPCA(概率主成分分析)
- 基于低秩的概率主成分分析方法插补缺失值。
- 适用场景:MCAR/MAR。
- NIPALS(非线性迭代部分最小二乘法)
- 通过低秩矩阵分解迭代估算缺失值。
- 适用场景:MCAR/MAR。
- SVD(奇异值分解)
- 通过低秩 SVD 矩阵分解进行插补,利用数据的全局结构。
- 适用场景:MCAR/MAR。
- SVT(奇异值阈值化)
- 通过核范数最小化方法解决缺失值问题,适用于低秩数据。
- 适用场景:MCAR/MAR。
Local Similarity
局部相似性
- 样本最近邻(Sample-wise KNN)
- 基于样本间的局部相似性,通过 kk-最近邻样本的加权平均值插补缺失值。
- 适用场景:MAR(随机缺失)。
- 蛋白质最近邻(Protein-wise KNN)
- 基于蛋白质之间的相似性,通过 kk-最近邻蛋白质的加权平均插补缺失值。
- 适用场景:MAR(随机缺失)。
- MNAR: 非随机缺失
- 缺失值与未观测的数据本身有关,例如“半最小值法”。
- MCAR: 完全随机缺失
- 缺失值与观测数据无关,例如 PPCA、SVD、SVT 等矩阵分解方法。
- MAR: 随机缺失
- 缺失值与观测数据有关,但与未观测数据无关,例如 KNN 方法。
Single cell RNA-seq
- MAGIC
- scImpute
- SAVER
- ALRA
- SAUCIE
MAGIC
1. 原始数据 (Original Data)
- 输入是单细胞转录组的原始基因表达矩阵。
- 数据特点:由于技术限制,数据通常稀疏且有噪声。
- 可视化:点图展示了细胞在一个隐空间中的分布。
2. 计算距离矩阵 (Calculate Distances)
- 目的:计算细胞间的距离,基于基因表达值。
- 方法:
- 使用欧几里得距离或其他距离度量。
- 结果:
- 生成一个对称的距离矩阵,表示细胞两两之间的相似性。
- 点图进一步展示细胞在空间中的连接趋势。
3. 计算亲和矩阵 (Calculate Affinities)
- 目的:将距离矩阵转化为亲和度矩阵(Affinity Matrix)。
- 方法:
- 使用高斯核(Gaussian Kernel),将距离映射为亲和度。
- 公式:$$Affinity\ = exp (-\frac{Distance^{2}}{2\sigma^{2}}) $$
- 结果:
- 亲和度矩阵中,较小的距离对应高的亲和值,较大的距离对应低的亲和值。
4. 马尔科夫标准化 (Markov Normalization)
- 目的:对亲和度矩阵进行标准化,使其满足马尔科夫性质(每行和为1)。
- 方法:
- 对矩阵进行归一化,得到马尔科夫过渡矩阵。
- 结果:
- 生成的矩阵可用于模拟细胞间的扩散过程。
5. 马尔科夫矩阵指数化 (Exponentiate Markov Matrix)
- 目的:模拟扩散过程,扩散步数由参数 tt 决定。
- 方法:
- 对马尔科夫矩阵进行
次幂
- 对马尔科夫矩阵进行
- 效果:
- 增加扩散步数
可以加深数据的平滑程度,消除噪声。
- 增加扩散步数
6. 基因表达补全 (Impute Gene Expression)
- 计算:
- 将扩散后的马尔科夫矩阵
应用于原始数据矩阵 - 对补全后的数据进行重缩放。
- 将扩散后的马尔科夫矩阵
- 结果:
- 得到一个平滑且补全的基因表达矩阵,能够更好地反映生物学信号。
MAGIC 算法通过以下步骤实现数据补全:
- 计算距离。
- 转化为亲和度。
- 使用马尔科夫模型模拟数据扩散。
- 对基因表达进行平滑和补全。
这是一种有效的基于图的方法,可显著改善单细胞数据的解析质量。
Spatial Transcriptomics
- stDiff
- GNTD
stDiff
Matrix Dimension Reduction
Linear Methods
PCA
cMDS
Non-linear methods
- t-SNE
- UMAP
- KPCA
- mMDS
- Isomap
- LLE (Locally Linear Embedding)
- Laplacian Eigenmap
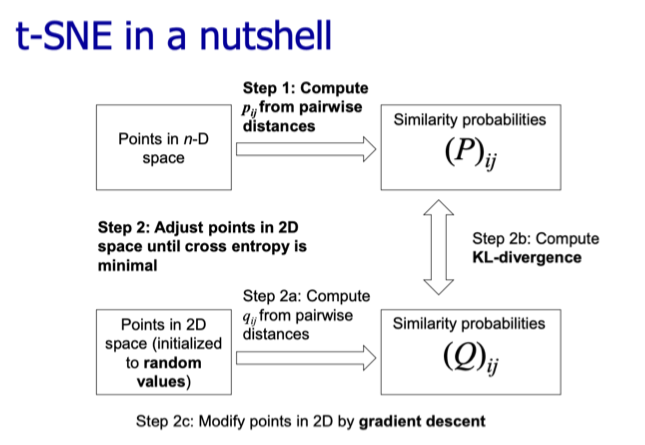
t-SNE
t-SNE(t-Distributed Stochastic Neighbor Embedding)是一种非线性降维技术,广泛用于高维数据的可视化。由 Laurens van der Maaten 和 Geoffrey Hinton 于 2008 年提出,t-SNE 能很好地保留数据的局部结构,因此特别适合于聚类或高维数据的可视化任务。
t-SNE 的基本原理
高维空间的相似度计算:
- 在高维空间中,t-SNE 用 高斯分布 表示点之间的相似性。
- 其中,
是点 的局部尺度参数,通过预定义的困惑度(Perplexity) 调整。
低维空间的相似度计算:
- 在低维空间中,点之间的相似性
用 学生分布(t-distribution) 表示:
这种分布的长尾特性使得低维空间更容易保留数据的局部结构。
优化目标:
- t-SNE 的目标是使高维空间的相似性
和低维空间的相似性 尽可能接近。使用 Kullback-Leibler 散度(KL 散度)作为优化目标:
- 通过梯度下降算法最小化这个损失函数。
t-SNE 的核心参数
困惑度(Perplexity):
- 用于平衡局部和全局结构的影响,通常值在 5 到 50 之间。
- 困惑度越高,模型关注的数据范围越大。
学习率(Learning Rate):
- 对优化过程的速度和质量有较大影响。
- 通常建议的值为 200,但可以根据数据调整。
迭代次数:
- t-SNE 通常需要 1000 次以上的迭代才能收敛。
优点:
- 非常适合用于高维数据的可视化。
- 能很好地保留数据的局部结构,易于发现数据的聚类特性。
局限性:
- 计算复杂度较高,不适合特别大规模的数据集。
- 不适合用作一般降维算法(例如 PCA 的替代)。
- 超参数(如困惑度、学习率)对结果影响较大,需要仔细调整。
- 结果的随机性强,运行多次可能得到不同的结果。
UMAP
t-SNE converts the distance between the two points into a probability in an ad hoc manner and attempts to preserve this probability
UMAP, starts with a totally different idea: To construct a manifold from only a sampling of the points on it
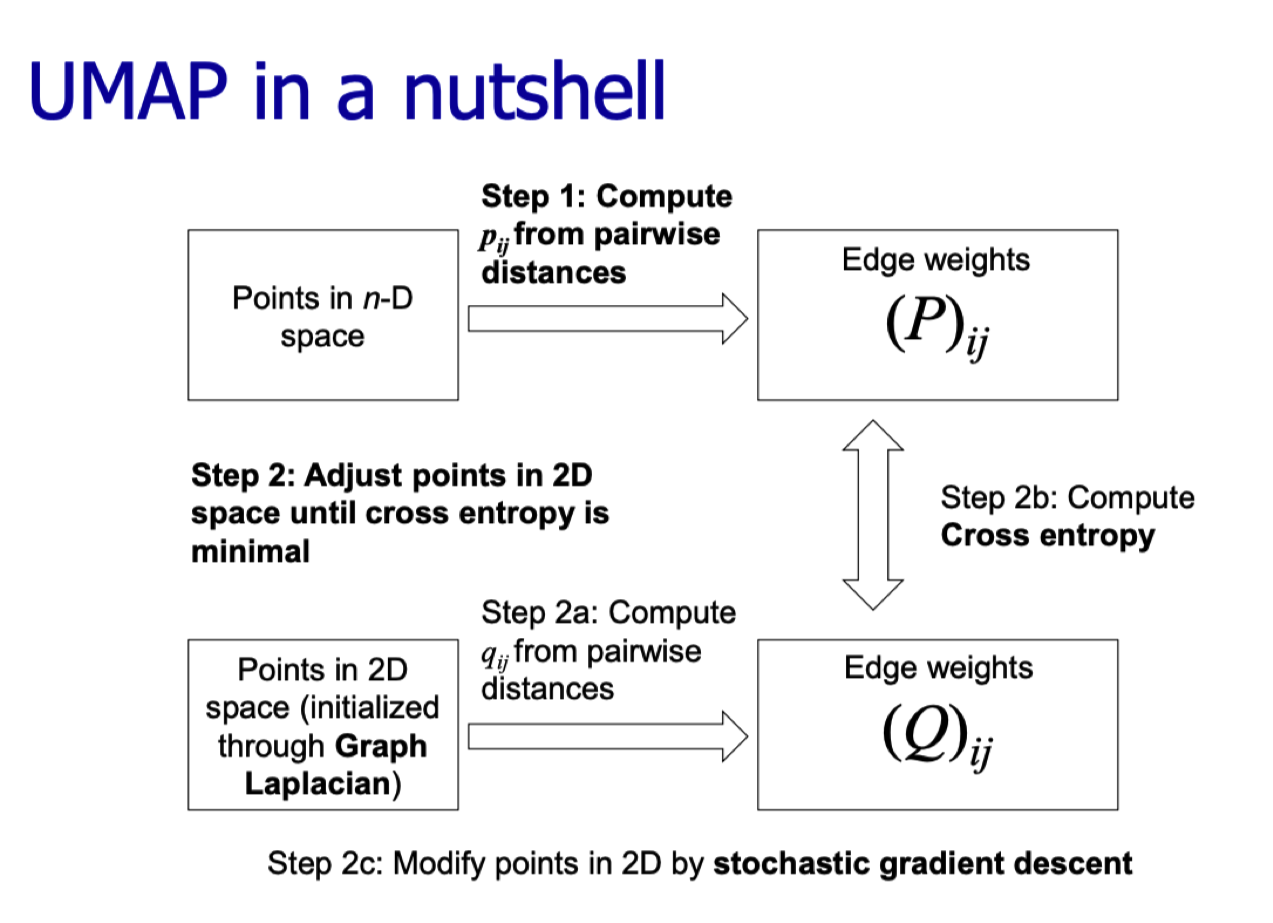
UMAP 的基本原理
UMAP 的理论基础是流形假设(Manifold Hypothesis),即高维数据在本质上分布在低维的流形上。UMAP 通过以下步骤实现降维:
构建高维空间的邻域图
- 计算邻居: 使用 k 近邻算法(k-NN)在高维空间中找到每个点的最近邻。
- 用户需设置
n_neighbors参数,决定邻域大小。
- 用户需设置
- 定义邻居相似度: 使用高斯分布计算邻域内点之间的相似性。
其中:
: 平滑参数,确保最近的邻居有较小的权重。 :局部尺度参数,通过优化确保邻域结构的一致性。 生成一个加权无向图,点之间的权重代表相似性。
将高维邻域图嵌入到低维空间
- 在低维空间中,点之间的距离用一种**单调下降的曲线(通常是倒数函数)**来衡量。
其中
- 优化目标是最小化高维邻域图和低维空间图之间的差异,使用交叉熵损失函数进行迭代优化。
优化过程
- UMAP 使用随机梯度下降(SGD)优化,将高维数据点映射到低维空间,同时保留高维数据的局部和全局结构。
- 与 t-SNE 不同,UMAP 的优化更高效,适用于大规模数据。
UMAP 的核心参数
n_neighbors(邻居数量)- 控制每个点的局部邻域大小,影响全局和局部结构的平衡。
- 小值:强调局部结构。
- 大值:保留更多全局结构。
min_dist(最小距离)- 控制嵌入空间中点之间的最小距离,影响聚类结果的紧密程度。
- 小值:点簇更紧凑,聚类更明显。
- 大值:点分布更稀疏。
n_components(降维后的维度)- 默认值为 2,适合可视化,但也可以设置为更高维度,用于特征提取或其他任务。
metric(距离度量)- 用于衡量高维空间中的点对相似性。默认是欧几里得距离,也支持其他距离(如曼哈顿距离、余弦相似度等)。
优点
- 速度快:UMAP 的计算效率比 t-SNE 高,适合大规模数据。
- 全局结构保留更好:UMAP 在降维时能同时保留局部和全局结构。
- 参数可调性强:可以通过
n_neighbors和min_dist调整对局部或全局结构的关注程度。 - 适用范围广:支持多种距离度量,可应用于不同类型的数据(如图像、文本、基因数据)。
局限性
- 参数较多,需要根据具体任务调整。
- 对嵌入的随机性敏感,不同运行可能会有略微不同的结果。
UMAP 与 t-SNE 的比较
| 特性 | UMAP | t-SNE |
|---|---|---|
| 速度 | 更快,适合大规模数据 | 较慢,特别是大数据集时 |
| 全局结构 | 更好地保留全局结构 | 更关注局部结构 |
| 参数调节 | 更灵活,支持多种距离度量 | 参数少但对结果影响较大 |
| 嵌入稳定性 | 嵌入结果更稳定 | 嵌入结果具有一定随机性 |
| 使用场景 | 数据探索、聚类、降维 | 数据可视化(主要用于 2D 或 3D) |